
Investigadores identifican una nueva variante genética responsable de una deficiencia severa de coenzima Q
Un grupo de investigación del Centro Andaluz de Biología del Desarrollo descubre una variante en el gen COQ4 que afecta la producción de energía celular. El estudio se centra en el caso de una paciente que falleció a los 16 meses de edad sin un diagnóstico certero, pese a presentar síntomas compatibles con una enfermedad mitocondrial. Este hallazgo pone en evidencia la necesidad de analizar regiones del ADN que tradicionalmente no se estudian en los análisis genéticos estándar.
Fuente: Universidad Pablo de Olavide
Las enfermedades raras afectan a millones de personas en todo el mundo, pero su diagnóstico sigue siendo un desafío debido a su baja prevalencia y la falta de conocimiento sobre sus causas genéticas. Un equipo de investigación del Centro Andaluz de Biología del Desarrollo (CABD) ha logrado identificar y validar una nueva variante genética en el gen COQ4, responsable de una deficiencia severa de coenzima Q (CoQ), un lípido esencial para la producción de energía en nuestras células.
Este hallazgo, publicado en la revista Molecular Genetics and Metabolism Reports, no solo amplía el conocimiento sobre estas enfermedades mitocondriales, sino que también pone en evidencia la necesidad de analizar regiones del ADN que tradicionalmente no se estudian en los análisis genéticos estándar.

La investigadora Gloria Brea observando células a través de microscopía.
El estudio se centra en el caso de una paciente que falleció a los 16 meses de edad sin un diagnóstico certero, pese a presentar síntomas compatibles con una enfermedad mitocondrial. Aunque se le realizó un análisis de exoma, una técnica que estudia únicamente las regiones codificantes del ADN, los resultados no ofrecieron una explicación clara para su enfermedad.
Sin embargo, un análisis más profundo permitió detectar un cambio en una región intrónica del gen COQ4. Aunque esta zona del ADN no codifica directamente proteínas, la mutación alteraba el proceso de maduración del ARN mensajero, lo que resultaba en una reducción drástica de la cantidad de proteína funcional producida por uno de los alelos del gen.
El otro alelo de COQ4 presentaba un cambio ya descrito previamente que generaba una proteína truncada y no funcional. Como consecuencia, la paciente no producía suficiente proteína COQ4, lo que afectaba la síntesis de coenzima Q, una molécula fundamental para el funcionamiento de la cadena de transporte de electrones mitocondrial y la producción de energía celular. Esta deficiencia energética tuvo consecuencias clínicas graves y finalmente resultó en un desenlace fatal.
“Este estudio ha permitido ampliar el catálogo de variantes patogénicas de COQ4 y refuerza la importancia de investigar regiones no codificantes del ADN, que podrían ser clave en muchos casos de enfermedades raras sin diagnóstico”, indica Gloria Brea, profesora del Área de Biología Celular de la Universidad Pablo de Olavide e investigadora principal del grupo ‘Bases moleculares de la deficiencia de Coenzima Q y bioenergética del desarrollo’ del CABD, centro mixto del Consejo Superior de Investigaciones Científicas (CSIC), la Universidad Pablo de Olavide y la Junta de Andalucía.
Esperanza para las familias
Desde 2015, cuando el equipo de investigación participó en la identificación de los primeros casos de variantes patogénicas en COQ4, solo se han descrito unas pocas decenas de pacientes en todo el mundo con esta deficiencia primaria de coenzima Q.
El diagnóstico de la paciente, aunque llegó tarde para un posible tratamiento, tuvo un impacto profundo en su familia. “En enfermedades raras, el acceso a un diagnóstico puede llevar hasta cinco años de media, lo que genera incertidumbre, ansiedad y dificultades para acceder a cuidados específicos. Saber con certeza qué causó la enfermedad de su hija permitió a los padres cerrar una etapa de incertidumbre y, además, tomar decisiones informadas sobre su futuro”, explica la investigadora de la UPO.
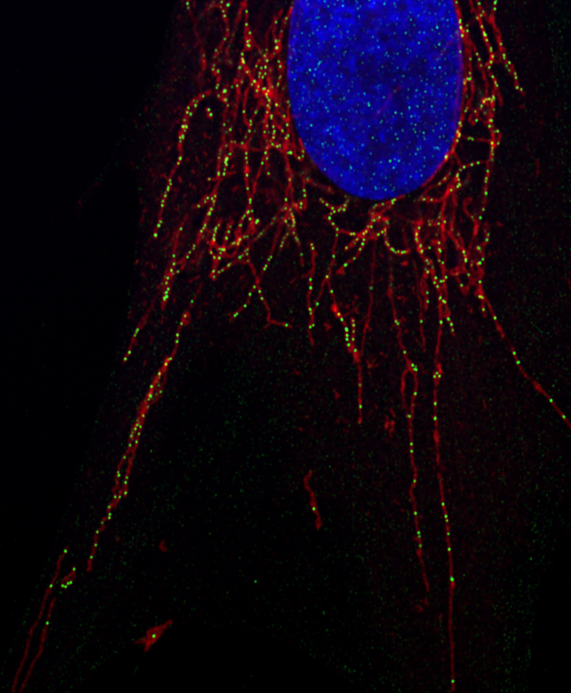
Imagen de una célula realizada con un microscopio confocal de súper resolución. Imagen: Ángela Moltó Domínguez.
Gracias al diagnóstico molecular, la familia pudo acceder al consejo genético apropiado, lo que les permitió tener dos hijos sanos. Para ellos, este avance supuso un cambio de vida radical, evitando que la enfermedad volviera a afectar a su descendencia.
“Este caso pone de manifiesto una realidad común en el ámbito de las enfermedades minoritarias: cada diagnóstico es una victoria, no solo para la comunidad científica, sino también para las familias que buscan respuestas”, declara María Alcázar-Fabra, autora principal del estudio.
Modelos animales y nuevas estrategias terapéuticas
El equipo de investigación lleva años estudiando las deficiencias primarias de coenzima Q y trabajando en su diagnóstico molecular. Sin embargo, debido a la baja prevalencia de la enfermedad, es difícil obtener suficientes datos clínicos para avanzar en su conocimiento y tratamiento.

María Alcázar-Fabra, primera autora del estudio.
Para superar esta limitación, el grupo ha obtenido financiación estatal para desarrollar modelos de pez cebra que porten las mutaciones identificadas en pacientes. Estos modelos permitirán estudiar la enfermedad desde sus primeras etapas de desarrollo, arrojando luz sobre cómo se origina y progresa.
Además, estos modelos servirán como plataforma para el cribado de fármacos, lo que podría abrir la puerta a nuevas estrategias terapéuticas. Aunque hoy en día no existe un tratamiento efectivo para la deficiencia de coenzima Q causada por mutaciones en COQ4, la posibilidad de probar distintos compuestos en un modelo animal acelerará la búsqueda de soluciones.
“Este trabajo no solo amplía nuestro conocimiento sobre las enfermedades mitocondriales raras, sino que también subraya la importancia de la investigación en genética y el impacto que puede tener en la vida de los pacientes y sus familias”, declara Gloria Brea.
Referencia:
Alcázar-Fabra et al. ‘Identification of a new COQ4 spliceogenic variant causing severe primary coenzyme Q deficiency’, Molecular Genetics and Metabolism Reports 42 (2025).
Últimas publicaciones

Un equipo de investigación de la Universidad de Málaga ha diseñado una estrategia de control para motores utilizados en movilidad eléctrica y aeronáutica. Esta técnica mejora la calidad de la corriente y predice el funcionamiento más adecuado, optimizando su eficiencia de consumo y respondiendo ante posibles fallos.

La comunidad tiene la oportunidad de realizar una evaluación previa este domingo para estimar la logística y seguridad necesarias de cara al evento astronómico que será visible en 115 localidades de Almería, Cádiz, Granada y Málaga.
Sigue leyendo
El programa de ocio estival de la Fundación Descubre estará presente en localidades de las provincias de Huelva, Sevilla, Málaga, Granada, Jaén y Almería.
Sigue leyendo