
Historia genómica del coronavirus en España, del enlace con Kazajistán al estigma de los temporeros
Un grupo antiguo del SARS-CoV-2 dominó en el confinamiento español de la primera oleada, ¿tuvo mayor virulencia? No. Se ha dicho que Aragón y Cataluña fueron foco de una nueva cepa por toda Europa durante el verano, ¿está ya probado? Tampoco. Las autoras de los últimos estudios genéticos validados sobre la pandemia en nuestro país y la directora del Instituto de Salud Carlos III aclaran dudas.
Fuente: Agencia SINC
¿Cómo llegó el coronavirus a España? ¿Cómo se extendió por nuestro territorio? La respuesta a estas preguntas está en los aproximadamente 30.000 nucleótidos que forman la cadena de ARN del SARS-CoV-2. Estas piezas no son siempre idénticas, sino que varían entre pacientes y a lo largo del tiempo debido a pequeños fallos que se producen durante la multiplicación del virus. La secuenciación de miles de muestras de pacientes permite reconstruir brotes y desvelar la naturaleza del patógeno. Conforme este puzle genómico se completa, aumenta nuestro conocimiento para combatir la pandemia.

Turista en España durante la pandemia. / Adobe Stock
Hacemos un repaso a la reconstrucción de este puzle y aprovechamos para desmentir algunas ideas erróneas surgidas de la interpretación de estudios no validados.
El extraño coronavirus de la primera ola
Un estudio del Centro Europeo para la Prevención y Control de Enfermedades publicado en la revista Eurosurveillance en el mes de agosto analizó las 39.000 secuencias disponibles hasta el 10 de julio, recogidas en 35 países de la región europea de la OMS. Su objetivo era estudiar la distribución geográfica y temporal de los distintos grupos genómicos (del inglés, clade) del coronavirus.
Estas pequeñas variaciones del SARS-CoV-2 provienen de un mismo ancestro, lo que permite retroceder en el tiempo para saber lo que pasó en el continente durante las primeras semanas y meses de pandemia. Así, España destaca en los resultados del trabajo por lo raro de los grupos genómicos observados en las aproximadamente 1.800 secuencias de la primera ola analizadas en nuestro país.
“El clado 19B/S ha sido muy raro, excepto en España y Kazajistán”, mientras que “las frecuencias fueron variadas en la mayoría de territorios”. Junto a este, nuestro país mostró un segundo grupo genómico, el 20A/G, “y muy poco de los otros”, según escriben los autores. En otras palabras, la diversidad de los coronavirus españoles fue escasa durante los inicios de la pandemia.
“El coronavirus que circuló en la primera ola en España pertenece a grupos antiguos, cuyas secuencias son de las primeras que salieron de Asia”, explica a SINC la investigadora del Instituto de Salud Carlos III (ISCIII) y coautora del estudio, Inmaculada Casas. Es una situación que se ve en todas las Comunidades Autónomas: “Más o menos tienen el mismo porcentaje [de estas variantes], que es muy alto si se compara con el resto de países europeos”.
Antes de continuar, conviene aclarar que en el contexto del nuevo coronavirus no se puede hablar de cepas (como las de la gripe), sino de variantes. Casas explica el motivo, que no es baladí: “Hay tan poca variedad en el coronavirus que se habla ‘grupos genómicos’, clados. Y dentro de cada grupo hay linajes”.
Atrapados en una primavera confinada
España fue uno de los primeros países europeos en los que la incidencia del coronavirus empezó a destacar. ¿A qué se debe esta falta de variación? “Las medidas de contingencia que se adoptaron para frenar la expansión del virus fueron de las más agresivas de Europa”, comenta Casas. “En febrero casi no había casos, solo importados, y cuando empezó a circular, al estar todo el país confinado y cerrado, no tuvimos una importación de nuevos grupos genómicos que estaban circulando en otros territorios”.
Como en El mundo perdido de Arthur Conan Doyle, los coronavirus españoles quedaron atrapados en el tiempo durante la primavera. “Los que se quedaron al principio de la pandemia, los más antiguos, estuvieron circulando más tiempo, a diferencia de otros países donde sí hubo una entrada de otros [grupos genómicos de SARS-CoV-2] porque las medidas de restricción no fueron tan estrictas”.
Es importante señalar que este trabajo es una fotografía de los primeros virus que circularon en marzo y abril, durante la implementación de las medidas de aislamiento, por lo que el escenario descrito en el estudio ya ha variado. “El objetivo era mirar lo que había en la base de datos de secuencias al inicio de la pandemia en los distintos países”, explica Casas. “Ahora empezamos a tener las secuencias de lo ocurrido en verano, pero esos virus ya no son de esos linajes antiguos, sobre todo con las introducciones que ha habido con el turismo y la movilidad”.
“El grupo inicial es el 19S [el número hace referencia al año 2019, cuando empieza la nomenclatura]. Todos los virus de Wuhan y de los países que empezaron entran dentro de ese linaje S, que fue exportado a Alemania y demás lugares”. También a España. “Estaban en China, evolucionaron un poco, llegaron aquí y con nuestras medidas de contención siguieron circulando”, añade Casas. Con la nueva normalidad del verano, el virus del grupo 19 desapareció para siempre: “Ahora son más parecidos a los del año 2020”.
El misterio de Kazajistán… e Italia
El lector quizá se pregunte a qué se debe la conexión con Kazajistán. Esa pregunta, por desgracia, quedará sin resolver. La investigadora del ISCIII y coautora del estudio María Iglesias sugiere que la explicación pueda estar en el “sesgo en la elección de muestras” que se secuenciaron allí.
Es algo que también señala el estudio: “En aquellos países con pocas secuencias, la distribución de los grupos genómicos puede ser poco fiable y puede haber un sesgo hacia aquellos que dominaron en diferentes fases de la pandemia según las estrategias de muestreo de cada momento y el tiempo en el envío de las secuencias”. Casas aclara que los territorios con menos recursos “no tienen secuencias ni un biobanco que guarde todas las muestras de la pandemia” como sucede en nuestro país.
Iglesias rechaza que este tipo de sesgos haya afectado a los resultados españoles: “En España esta posibilidad no existe porque hay muchas muestras analizadas a lo largo del tiempo”. Sí considera “curioso” que Italia no tenga un perfil similar al de España y sí muestre variabilidad en sus grupos genómicos. “Quizá, al ser el origen del brote en Europa y tener mucho flujo turístico, tuviera más virus diferentes circulando y por eso vemos perfiles diferentes”.
Entró muchas veces desde enero, pero no cuajó
Un estudio de Iglesias, compartido en forma de preprint en abril y publicado hace unos días en la revista Journal of Virology coincide con el trabajo de Eurosurveillance: el virus tuvo varios focos de entrada en España, pero solo unas pocas de estas ascuas prendieron y se extendieron, lo que provocó un efecto fundador.
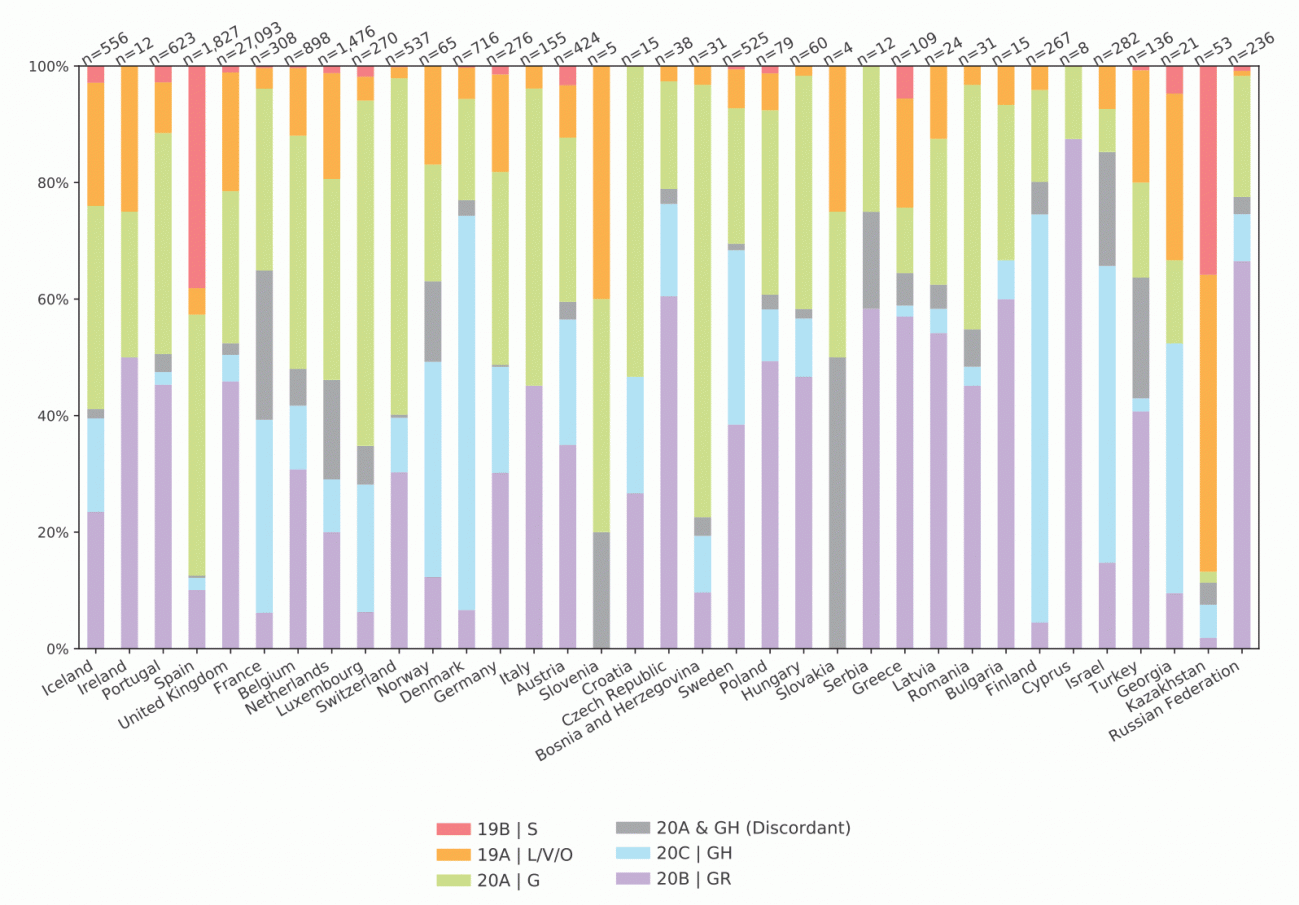
Distribución de los clados de SARS-CoV-2 entre países de la región europea de la OMS, basados en muestras secuenciadas entre febrero y julio de 2020. En la cuarta columna por la izquierda se observa el predominio de la variante 19 B/S en España. Fuente: Eurosurveillance.
“Es algo que ha pasado en toda Europa: tener una única entrada es algo que ya no sucede”, explica Iglesias en referencia a la alta conexión que existe hoy entre países y territorios. “Normalmente hay varias y en algunos sitios, donde la transmisión es más fuerte, podemos trazar mejor el origen de esos linajes y su región geográfica”.
Es lo que hizo Iglesias, que encontró 62 variantes de SARS-CoV-2 introducidas en España, de las que cuatro generaron transmisión local entre finales de enero y principios de febrero. Así, la variante 19B se convirtió en la reina de la jungla durante un tiempo.
Este reinado no duró mucho: las variantes más frecuentes al inicio fueron sustituidas por otras con una mutación en la proteína de la espícula del virus que hoy domina en todo el mundo: D614G. El estudio de Iglesias también concluyó, mediante experimentos in vitro, que esta ventaja no es casual sino fruto de una mayor capacidad infectiva.
A la izquierda, el origen geográfico de las 61 muestras secuenciadas para el trabajo. A la derecha, árbol filogenético que muestra los clados de 2019 en naranja y los de 2020 en verde. En rosa, las 62 introducciones independientes observadas en España. También se muestran, en letra pequeña, los cuatro clústeres españoles. Fuente: Journal of Virology
Cuidado con las malinterpretaciones genómicas
La idea de un coronavirus raro, único y antiguo circulando por España en los primeros meses de la pandemia resulta tan interesante como atractiva para hacer juicios precipitados. Casas niega rotundamente que esto pueda tener relación alguna con la letalidad observada en marzo, una mejor —o peor— transmisión del virus en nuestro país o una mayor susceptibilidad a reinfecciones futuras.
“La letalidad es una forma de interpretar los datos”, advierte Casas. “No tiene nada que ver con el virus, sino que es una cuestión de cómo medir variables epidemiológicas”.
Así, en la primera ola la mayoría de positivos eran pacientes que ingresaban graves, mientras que en verano se incrementaron los diagnósticos para detectar casos leves, lo que disminuyó la proporción de fallecidos. Por todo ello considera “erróneas” las ideas que sugieren que el virus es hoy menos virulento.
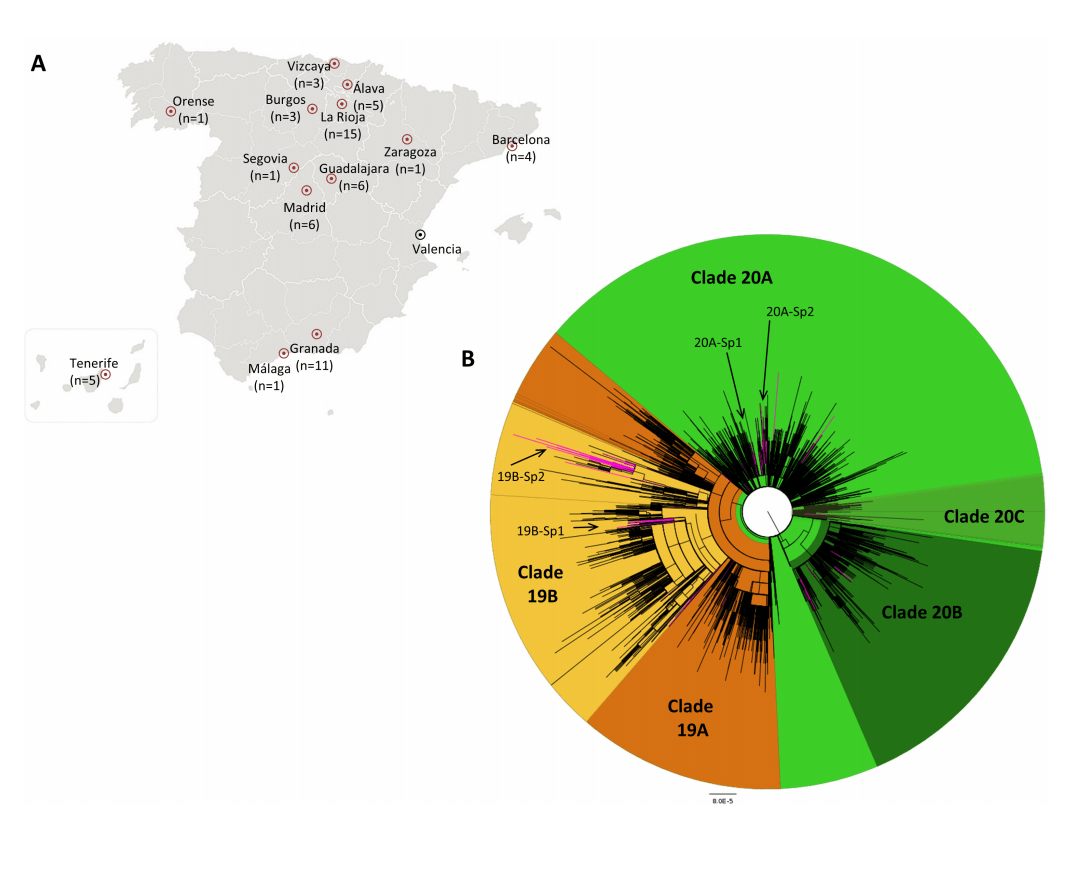
A la izquierda, el origen geográfico de las 61 muestras secuenciadas para el trabajo. A la derecha, árbol filogenético que muestra los clados de 2019 en naranja y los de 2020 en verde. En rosa, las 62 introducciones independientes observadas en España. También se muestran, en letra pequeña, los cuatro clústeres españoles. Fuente: Journal of Virology
Tampoco cree que se puedan hacer extrapolaciones sobre una mayor transmisión. De hecho, la mutación en la proteína de la espícula D614G culpable de una mejora en la transmisibilidad del SARS-CoV-2 es más reciente que la variante 19B/S que predominó en España por entonces. Casas matiza que esto no quiere decir que las versiones anteriores del coronavirus fueran incapaces de contagiar.
“Cuando se estudia la espícula del virus se ve que es un supervirus muy adaptado al ser humano desde el primer momento. No le hacen falta mutaciones para adaptarse mejor y me atrevo a decir que cualquiera que se produzca, tanto en la espícula como en otras regiones genómicas, va a ir en su contra”, dice la investigadora. “Ahora mismo está muy adaptado al receptor del ser humano y de los mamíferos ACE2 y cualquier cambio le podría salir mal”.
Casas asegura que las progenies virales que sobreviven y circulan hoy son las que están mejor adaptadas, pero no considera alarmante que en 30.000 nucleótidos haya una mutación en la proteína de la espícula. “Para los que trabajamos con gripe esto no es nada”, comenta, en relación con la alta tasa de variabilidad de este otro virus.
¿Podrían los españoles que estuvieron expuestos al coronavirus de marzo ser más propensos a una reinfección futura? Casas también lo niega. “El virus es idéntico. Aunque tenga varias mutaciones, los anticuerpos que reconocen la espícula de los grupos iniciales siguen reconociendo los virus de octubre a la perfección”. En otras palabras, el virus no se “escapa” de nuestro sistema inmunitario con tanta facilidad como el de la gripe.
Estos tres puntos pueden resumirse en uno solo: “El virus es muy parecido desde el principio, no puede decirse que los grupos antiguos sean tremendamente diferentes a los de ahora, y menos en términos de control inmunitario”.
Divulgación responsable y resultados preliminares
La directora del ISCIII, Raquel Yotti, iba un paso más allá en un tuit reciente en el que advertía contra otro tipo de malinterpretaciones, fruto de sacar “conclusiones contundentes” a partir de “estudios aún no revisados”. Mencionaba también la “irresponsabilidad” de que estos trabajos preliminares puedan servir [como sucedió en algunos medios] para “culpar, estigmatizar o reforzar estereotipos”.
Las 30.000 letras que forman el coronavirus pueden revelarnos mucho sobre su origen, pero también señalar a colectivos desfavorecidos y a países enteros. Por ejemplo, un preprint aún no revisado, cuyas conclusiones aclaramos en este artículo, sugiere que la variante del SARS-CoV-2 que domina hoy Europa surgió entre los temporeros de Aragón y Cataluña. Yotti cree que hay que apostar por los matices a la hora de comunicar trabajos en medios: “Es muy raro que un artículo científico haga afirmaciones contundentes, pero con la pandemia hay mucha atención mediática”.
Esto provoca que en los medios “mensajes científicos se simplifiquen y se corra el riesgo de perder rigor y enviar mensajes erróneos que puedan ser muy perjudiciales”, continúa Yotti. “[En el ISCIII] intentamos trasladar a nuestros investigadores esa precaución y sentido de la responsabilidad con todo lo que no está publicado y que se encuentra en el ámbito de las hipótesis, las ideas y las prepublicaciones”.
Por el contrario, “cuando se lanza una noticia al mismo tiempo que una prepublicación”, no se da tiempo a que otros investigadores analicen los datos. En el caso del reciente trabajo que sitúa los brotes entre temporeros como origen de la nueva variante, Yotti cree que “hay ciertas cuestiones aún por dirimir”, pide cautela y lamenta que tras su difusión mediática quede la noción de que “España está distribuyendo el virus por el mundo entero”.
Conocer al enemigo para saber cómo atacarlo
Las técnicas de secuenciación masiva se han implementado por todo el mundo a gran velocidad, en un esfuerzo por aumentar el conocimiento genómico del coronavirus. ¿Cómo ayudan este tipo de estudios en nuestra lucha contra la covid-19? Casas explica que resulta muy útil conocer si un virus cambia con rapidez o no, tanto para el control de la pandemia como para el tratamiento de los pacientes y el desarrollo de vacunas.
“La formación de resistencias es algo muy preocupante a la hora de tratar las infecciones virales”, asegura. Algunos virus intentan “escapar” del tratamiento dado al paciente, porque su alta tasa de variación genera progenies resistentes. Sin embargo, en patógenos que cambian menos, como el SARS-CoV-2, “se pueden usar mayores dosis de antivirales y moléculas más agresivas” con menos miedo a que esto pase.
El diseño de los tratamientos no es el único que se ve afectado por la variabilidad del coronavirus. “Es crucial saber la tasa de variación del SARS-CoV-2 en el diseño de las vacunas”, dice Casas. Por este motivo, defiende que este tipo de estudios genómicos “aportarán muchísima luz” a la hora de “evaluar la eficacia de las vacunas en el futuro”.
“Cuando se dirigen los anticuerpos contra un virus vacunal lo normal en un virus de ARN [como el coronavirus] es que intente escapar de ese control”, aclara Casas. Esto incluye a virus cuya tasa de mutación es baja en comparación con la de la gripe, como sucede con el SARS-CoV-2. “Estos estudios nos permitirán evaluar esas vacunas”. En otras palabras, prever cómo reaccionará el patógeno ante esa amenaza.
A todo esto hay que sumar que “es muy interesante” saber la variabilidad y secuencias de los virus que circulan en ciudades y brotes para “poder atajarlo e instaurar medidas de control específicas y adecuadas”. A fin de cuentas, “no hay un tratamiento general para las enfermedades virales. Cada una es un mundo y tiene sus características”.
Últimas publicaciones

Un equipo de investigación de la Universidad de Cádiz ha identificado estas partículas plásticas en diez playas de esta ínsula antártica, una de las zonas más aisladas del mundo. Los resultados sirven para generar un punto de referencia inicial que permita evaluar en el futuro si la contaminación aumenta.
Sigue leyendo
Esta nueva edición se celebrará el próximo viernes 17 de abril en el pasillo central de la Universidad de Almería bajo el lema ‘Una ventana abierta a la Ciencia’. Contará con más de 40 proyectos de investigación, 31 centros educativos participantes, así como unos 800 alumnos investigadores. Habrá exposiciones, talleres, conferencias y Café con Ciencia.
La Universidad de Sevilla se encarga de la investigación, impulsada por Adhera Health, en el diseño y la evaluación para mejorar el autocuidado y acompañar a pacientes, familias y profesionales en el paso de la atención pediátrica a la adulta.
