
Obtienen la secuencia del 100 % del genoma del virus de la viruela del mono
Un equipo del Laboratorio de Arbovirus y de las Unidades de Genómica y Bioinformática del Instituto de Salud Carlos III ha logrado la secuenciación completa del virus de la viruela del mono, un logro que permitirá hacer análisis filogenéticos más avanzados para obtener datos sobre su comportamiento y comprender mejor su origen, circulación y difusión.
Fuente: Agencia SINC
Investigadores del Instituto de Salud Carlos III (ISCIII) han logrado el primer borrador de secuencia completa del virus de la viruela del mono, obtenido del análisis genómico de muestras de 23 pacientes. Esta labor de secuenciación masiva permite confirmar que es la variedad de África Occidental la causante de este brote.
Adicionalmente, la secuenciación ha alcanzado una cobertura del 100 % de los 190.000 pares de bases del genoma de este virus, lo cual abre la posibilidad de estudios filogenéticos más avanzados que permitirán obtener información adicional sobre su comportamiento y comprender mejor su origen, circulación y difusión. Se trata de una de las secuencias más completas obtenidas hasta el momento.
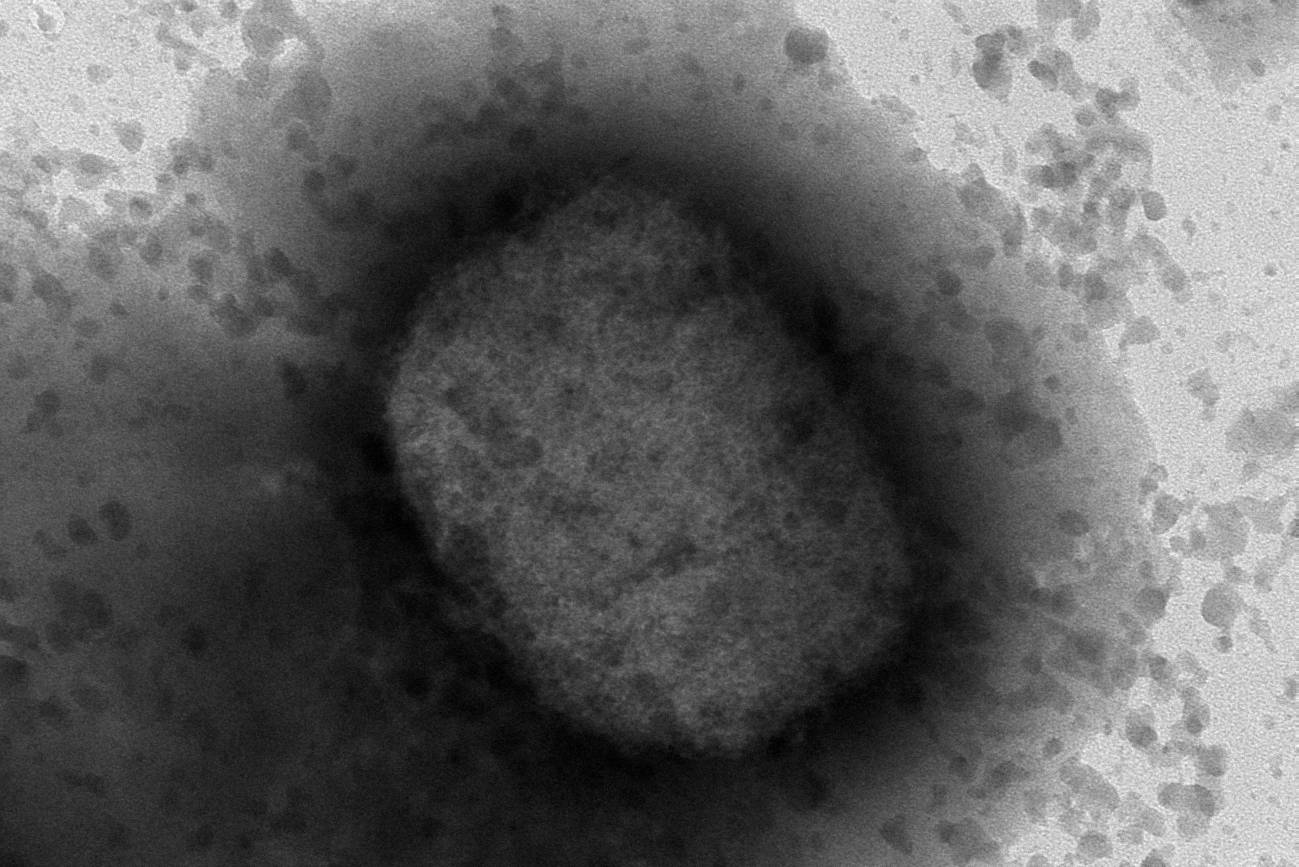
Imagen Mokeypox microscopía electrónica. / ISCIII
La secuenciación llevada a cabo a través del Laboratorio de Arbovirus del ISCIII, en conjunto con las unidades de Genómica y Bioinformática del, ha contado con las referencias publicadas en los últimos días por otros países (Bélgica, Alemania, Portugal y EEUU), y se ha basado en una tecnología genómica de nueva generación mapeada contra genoma de referencia, a lo que se ha sumado un análisis complementario de las muestras mediante una técnica conocida como ensamblado de novo. Las secuencias en crudo se terminaron de obtener el pasado lunes por la noche y el análisis de computación se ha realizado en las últimas 36 horas.
Los resultados señalan que las muestras secuenciadas parecen pertenecer al mismo brote detectado en otros países europeos, ya que los genomas obtenidos apenas difieren de los ya secuenciados en otros países; en concreto, el análisis realizado en el ISCIII concluye que hay menos de 5 SNP -secuencias genéticas denominadas cambios de nucleótidos sencillos- de diferencia respecto a la secuenciación llevada a cabo en Alemania.
La secuenciación confirma que el virus de la viruela de los monos del brote que se está produciendo en España es del clado de África Occidental, que es el de menor virulencia entre los conocidos y el que se ha identificado por el momento en la mayoría de los países fuera de África implicados en este brote. Los clados son grupos filogenéticos que definen la evolución biológica de un organismo, que explican cómo actúa y se comporta, y en ellos se pueden observan las diferencias genéticas de los virus circulantes.
Tras obtener la secuencia completa del virus, varios equipos de investigadoras del ISCIII están llevando a cabo análisis filogenéticos para conocer la relación entre las muestras españolas y las de otros países.
La información obtenida se enfrentará a las ya conocidas y depositadas en bases de datos internacionales para evaluar el grado de identidad y, en su caso, la localización de las diferencias que pudieran existir entre la secuencia española y los demás datos internacionales. Todo ello permitirá realizar los estudios de trazabilidad del brote, así como identificar potencialmente su origen.
Últimas publicaciones

El próximo 12 de agosto, al atardecer, las miradas de curiosos y aficionados a la astronomía apuntarán al cielo. El primero de los tres eclipses que se sucederán en 2026, 2027 y 2028 se iniciará a las 19:39, y llegará a su fase máxima hacia las 20:30, para finalizar entre las 21:15 y 21:25, dependiendo de la zona dónde se observe. En Andalucía se observará de forma parcial, y aunque el Sol no esté totalmente oculto, los expertos recomiendan protección ocular con gafas homologadas, evitar trucos caseros y poco efectivos como gafas de sol convencionales, radiografías, CD o cristales ahumados, ir debidamente equipados con agua y ropa de abrigo, así como escoger lugares abiertos y seguros, siempre mirando al horizonte occidental despejado.

Investigadores de las universidades de Almería y Granada han identificado en varios puntos cercanos a la capital almeriense afloramientos de origen marino correspondientes a la época geológica previa en la que el Mediterráneo se secó casi por completo. En estos arrecifes formados a casi 40 metros bajo el nivel del mar, la transparencia del agua en ese entorno facilitó el crecimiento de corales de lado a lado. Ahora aportan pistas para reconstruir la historia climática del pasado.

Un equipo de investigación de la Universidad de Málaga ha diseñado una estrategia de control para motores utilizados en movilidad eléctrica y aeronáutica. Esta técnica mejora la calidad de la corriente y predice el funcionamiento más adecuado, optimizando su eficiencia de consumo y respondiendo ante posibles fallos.
